
MOLECULAR MECHANISMS OF CHARCOT-MARIE-TOOTH DISEASE
Charcot-Marie-Tooth disease (CMT) is the most common inherited disorder of the peripheral nervous system, affecting up to one in 2,500 people. The disease is characterised by degeneration of the peripheral nerves in the feet, legs, arms and hands, causing muscle weakness, loss of limb function, and mobility impairments. There is no cure for CMT, and patients frequently suffer lifelong disabilities.
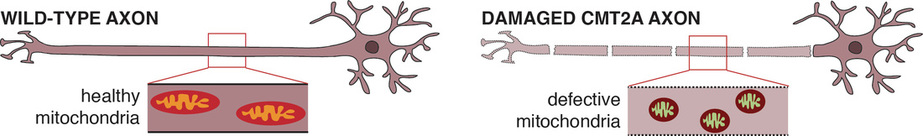
We model CMT in C. elegans in order to uncover novel information about how the disease develops, and therefore provide a better understanding of the disease. Our research focuses on axonal forms of CMT (classified as CMT2), and in particular on the most common axonal form, CMT2A, which is caused by mutations in Mitofusin 2, a protein critical for normal mitochondrial functioning.
Discovering effective therapeutics for CHARCOT-MARIE-TOOTH DISEASE
CMT is currently incurable and no effective pharmaceutical treatments have been developed. We are using our C. elegans models of disease to screen through thousands of compounds in order to identify potential therapeutics. Our C. elegans models permit rapid and cost-effective drug screening, and as most of the compounds we are testing have existing safety profiles in humans, this permits the rapid translation of our findings into the clinic. We aim to provide important insights into the mechanisms of action for lead compounds and to ascertain whether lead compounds are specifically effective against CMT2A, or whether they are more broadly effective against different subtypes of CMT.
DEFINING THE FUNCTION OF MITOFUSIN 2
In addition to modelling CMT2A, we are also investigating the function of the causative gene (MFN2). MFN2 is conserved in C. elegans, where it is known as fzo-1. We aim to uncover novel insights into the precise role of Mitofusin-2/FZO-1 in mitochondrial dynamics and to identify novel regulators of its activity. Mitofusin-2 is essential for normal mitochondrial health, and in addition to CMT2A is associated with other neurodegenerative (including Alzheimer's, Parkinson's, and Huntington's diseases), and non-neuronal conditions, including metabolic (diabetes and obesity) and vascular proliferative diseases. As such, our studies aim to fully characterise the role of protein that it vital for normal cellular function and that is linked to a variety of human disorders.
Representative Publications
Soh MS, Cheng X, Liu J, Neumann B (2019). Disruption of genes associated with Charcot-Marie-Tooth type 2 lead to common behavioural, cellular and molecular defects in Caenorhabditis elegans. bioRxiv 605584; doi: https://doi.org/10.1101/605584. Brief summary.
Byrne JJ, Soh MS*, Chandhok G*, Vijayaraghavan T, Teoh JS, Crawford S, Cobham AE, Yapa NMB, Mirth CK, Neumann B (2019). Disruption of mitochondrial dynamics affects behaviour and lifespan in Caenorhabditis elegans. Cellular and Molecular Life Sciences https://doi.org/10.1007/s00018-019-03024-5. Brief summary.
Chandhok S, Lazarou M, and Neumann B (2018). Structure, Function, and Regulation of Mitofusin-2 in Health and Disease. Biological Reviews - Review 93(2): 933-949. Journal link.

Individual muscle fibres of C. elegans visualised with a fluorescent protein